Volume 1, Issue 3
Predicting KEGG Orthologs Associated with Microbial Metabolism in Autotrophic Freshwater Microbes Using a Statistical Model
By Courtney-Grace Neizer

is a fourth-year honors student majoring in Computer Science with a concentration in Bioinformatics. Her research focuses on genomics and computational methods, with a focus on environmental exposures, especially concerning the impact of chronic conditions in low and middle-income countries. Her research advisors are Dr. Elaine Luo from the Department of Biological Sciences and Dr. Wenyu Gao from the Department of Mathematics and Statistics and the School of Data Science.
Abstract
Microbes play a crucial role in Earth’s biogeochemical cycles, yet linking microbial KEGG orthologs to carbon fixation remains challenging due to fragmented datasets and limitations in functional annotation. This study analyzed microbial DNA fragments from Siders Pond in Falmouth, Massachusetts, a salt-stratified meromictic lake. Microbial DNA fragments recovered through metagenomic sequencing of environmental samples were linked to microbial activity to carbon cycling using the DNA-stable isotope probing (DNA-SIP) methods and the important features selected using the LASSO regression statistical model. Environmental samples were incubated with 12C or 13C labeled dissolved inorganic carbon to track microbial carbon incorporation, followed by metagenomic sequencing. Contigs were annotated using both the Protein Families Database (PFAM) and the KEGG Orthology (KO) database, with a bit score threshold of >30, and were linked to excess atom fraction (EAF) values representing microbial carbon assimilation. While both annotation sources were utilized, a greater number of KEGG (Kyoto Encyclopedia of Genes and Genomes) orthologs were identified in this specific dataset, guiding the focus of the analysis. LASSO regression identified key KEGG orthologs potentially involved in carbon cycling. The approach resulted in identifying acyl-CoA synthetase (K00142), BamB – Outer membrane assembly (K17713), glucose-fructose oxidoreductase (K00118), and 23S rRNA pseudouridine2604 synthase (K06182), as key features associated with microbial metabolic processes potentially influencing carbon cycling. Additionally, a domain within hydrazine synthase plays a role in anaerobic ammonium oxidation (PF18582), linking the nitrogen and carbon cycles by converting ammonium and nitric oxide into hydrazine. This suggests a potential role for hydrazine synthesis in microbial carbon metabolism under anoxic conditions. It contributes to a better understanding of microbial roles in carbon cycling and explores new ways of using statistical models to study environmental systems. The findings could help expand knowledge on how microbes influence global carbon cycles. They highlight the potential to uncover novel carbon-fixing pathways, which are crucial for climate and sustainability research.
: Carbon Fixation, Chemosynthesis, Metagenomics, Functional Annotation, DNA-SIP, Excess Atom Fraction (EAF), LASSO regression.
Introduction
Microbes, tiny organisms found everywhere on Earth, play a vital role in maintaining a healthy environment. One of their key contributions is carbon cycling, the process of recycling carbon between the air, soil, and water [1], which supports life on Earth. These organisms can influence Earth’s climate by storing carbon in the ground and interacting with carbon in the atmosphere, influencing plant growth and climate balance [2].
However, while microbial roles in carbon cycling are well-established, our ability to predict which microbes are actively fixing carbon remains limited. In the past few decades, new technologies such as DNA sequencing and metagenomics have been widely used to understand the inner universe of these tiny creatures. Traditional genome-based approaches rely on the recovery and assembly of metagenome-assembled genomes (MAGs) and their microbial metabolism inferred using diverse biological databases able to classify and predict their ecological function and biological meaning. Meanwhile, as the diversity and functions of microbial genes and proteins are vast, existing databases often lack the information needed to fully characterize them, leaving many orthologs annotated as unknown or misclassified.
To improve biological classifications, new computational methods are being developed to facilitate the identification and classification of DNA sequences that were not totally recognized in the previous databases. The present study focuses on using an advanced statistical method to link KEGG orthologs associated with microbial metabolism to potential carbon cycling activity. Samples were collected from Siders Pond, a salt-stratified meromictic lake in Massachusetts, where unique microbial communities are capable of fixing carbon in the absence of sunlight. A key challenge in this research is that existing annotation databases, including PFAM and KEGG, sometimes lack the resolution to accurately classify microbial functions, leading to potential mis annotations. To overcome this limitation, this study integrates DNA-SIP with metagenomic sequencing, enabling the identification of microbial KEGG orthologs potentially involved in carbon fixation activity. This represents the first recorded instance of metagenomic-SIP being applied in a freshwater system, demonstrating its potential to validate functional predictions beyond database annotations.
Water samples were collected, filtered to isolate bacterial cells, and incubated using ¹³C (label) or 12C (control) to track carbon incorporation over time. The excess atom fraction (EAF) analysis was applied to measure the incorporation of isotopically labeled carbon (¹³C) into microbial genomes, allowing the identification of key microbial players in carbon fixation. By using this method, we were able to identify organisms performing chemosynthesis—carbon fixation without sunlight— through the incorporation of ¹³C revealing their ability to perform carbon fixation in a light-limited environment. By calculating EAF values for each DNA sequence (contig) obtained from metagenomic sequencing, the objective was to associate these values with specific contigs to infer the potential metabolic functions represented by KEGG orthologs, which may be linked to microbial carbon cycling.
To analyze this data, this study employs LASSO (Least Absolute Shrinkage and Selection Operator) regression, a statistical technique that identifies the most relevant KEGG orthologs from high-dimensional metagenomic data. LASSO was chosen because it effectively reduces large, complex datasets by selecting the most relevant features while minimizing noise. Previous studies have applied machine learning to metagenomic data [4], but few have directly linked EAF-based functional analysis to predictive models of carbon cycling.
By integrating metagenomics, EAF calculations, and statistical methods, this research aims to improve our understanding of microbial roles in carbon fixation and identifying of novel carbon-fixing pathways. Existing databases have limited ability to detect new pathways. To address this issue, this study seeks to fill that gap by uncovering previously unannotated KEGG orthologs linked to carbon cycling.
Materials and Methods
Sample Collection and DNA Processing
Environmental water samples were collected from Siders Pond, a meromictic lake in Massachusetts, where microbial communities engage in light-independent carbon fixation. Water samples were filtered to isolate bacterial cells, followed by a 7-day incubation to track carbon incorporation over time. After incubation, microbial DNA was extracted and sequenced, generating raw reads that were assembled into contigs, which are DNA fragments containing genomic information about microbial activity. The DNA assembly process moves from readings to contigs, scaffolds, and full genomes [17].
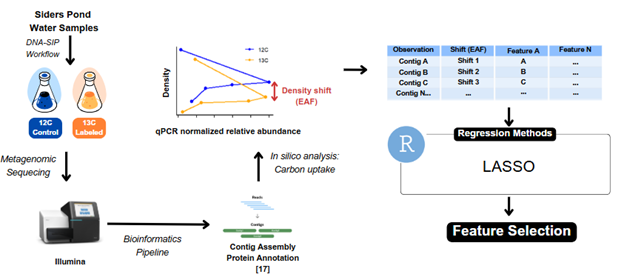
Figure 1. Water samples were collected from Siders Pond, a meromictic lake in Massachusetts. DNA-SIP was used to track microbial carbon fixation activity by incorporating either ¹³C-labeled or ¹²C-control dissolved inorganic carbon (DIC). DNA was sequenced, assembled into contigs, and analyzed for density shifts (EAF) to identify carbon fixation. The dataset containing EAF values and features was analyzed using LASSO regression in R programming language for statistical computing
To ensure data quality, sequencing data underwent preprocessing, including the removal of low-quality sequences to minimize errors. Contigs were annotated using the Protein Families (PFAM) database to identify protein domains, and sparsity in the dataset was addressed by replacing missing annotations with zeros [5]. KEGG Ortholog (KO) annotations were also included in the dataset and used to interpret microbial metabolic functions. Although the study initially aimed to focus on protein-level annotations, the final analysis emphasized KEGG orthologs due to their higher representation in this dataset, allowing for functional interpretation across gene, protein, and pathway levels.
Dataset Selection and Feature Processing
This study exclusively utilized the 5-kilobase (kb) contig dataset, as it was the primary dataset analyzed within the scope and timeline of the project. With its focus being on microbial sequences with annotated KEGG orthologs and functional domains associated with carbon cycling. To refine the dataset, features were filtered using a bit score threshold of ≥30, ensuring high-confidence annotations. Features with scores below this threshold were excluded to ensure high-confidence annotation [6,7]. Additionally, Excess Atom Fraction (EAF) analysis was incorporated to identify chemosynthetic microbial activity by tracking the incorporation of ¹³C-labeled carbon into microbial genomes.
Statistical Approach
To predict microbial contributions to carbon cycling, LASSO regression was applied. LASSO was selected for its ability to perform feature selection in high-dimensional datasets by identifying the most relevant KEGG orthologs while minimizing overfitting. The dataset was split into 80% training and 20% testing subsets, and cross-validation was performed to optimize model performance. The Root Mean Squared Error (RMSE) was calculated to assess the accuracy of predictions.
Dataset
RMSE
Training
0.0262900
Testing
0.0277889
Table 1. Root Mean Squared Error (RMSE) for LASSO Model. Representation of the RMSE values for the training and testing datasets, indicating the model’s predictive performance. Lower RMSE values suggest that the top 10 selected features, which included nine KEGG orthologs and one PFAM annotated protein domain, contribute meaningfully to carbon cycling predictions. However, further validation is necessary to confirm their ecological relevance. Visualization generated using R.
Data Analysis Workflow
The analytical workflow consisted of several steps. First, raw sequencing data were filtered, and missing values were addressed through preprocessing. Next, a bit score threshold was applied to the annotated dataset, followed by LASSO regression to identify significant microbial features associated with carbon cycling. The dataset was then divided into training and testing sets, and model performance was evaluated based on RMSE. The selected features included KEGG orthologs and one PFAM protein domain, which were further examined to explore their potential roles in microbial carbon fixation.
Computational Tools
All analyses were conducted using R, utilizing the glmnet package for LASSO regression and tidyverse for data preprocessing [14].
Data Availability
The dataset analyzed in this study consists of metagenomic sequencing data processed to extract contig-based features.
Results
This study identifies a subset of PFAM features that are potentially associated with microbial functions related to carbon cycling using EAF as a resource to measure the absorption of 13C by chemosynthetic microbes. The LASSO regression model selected these features based on the EAF measurement. The findings reinforce the potential of contig-based datasets for functional profiling by identifying microbial metabolic orthologs that may play critical roles in carbon cycling.
The bar plot (Figure 2) presents the top 10 microbial features selected by LASSO regression, ranked based on their association with excess atom fraction (EAF) values. The x-axis represents the coefficient values, indicating the relative contribution of each feature to the model’s predictions. The selected features include nine KEGG orthologs and one PFAM-annotated protein domain. Features with larger absolute coefficients have a stronger predictive relationship with microbial carbon cycling activity. In LASSO regression, features with larger absolute coefficients are more predictive within the model, as the method applies a penalty to less relevant variables, shrinking them to zero while retaining the most informative ones [18]. In this study, positive coefficients suggest a potential association with increased microbial carbon fixation, while negative coefficients may indicate an inverse relationship. However, as LASSO selects features based on statistical importance rather than direct biological causation, further validation is necessary to confirm functional relevance [18].
The y-axis lists the top 10 microbial features identified by LASSO regression, including nine KEGG orthologs and one PFAM domain (PF18582, hydrazine synthase subunit). These features were selected based on their association with excess atom fraction (EAF) values, indicating potential involvement in microbial carbon assimilation. While several of the identified orthologs have known roles in metabolism, membrane transport, and signaling, their specific contributions to carbon fixation remain to be experimentally validated [10]. Their selection by the model indicates that they were among the most predictive features in this dataset, but additional biological validation is required to determine whether they directly contribute to carbon cycling processes.
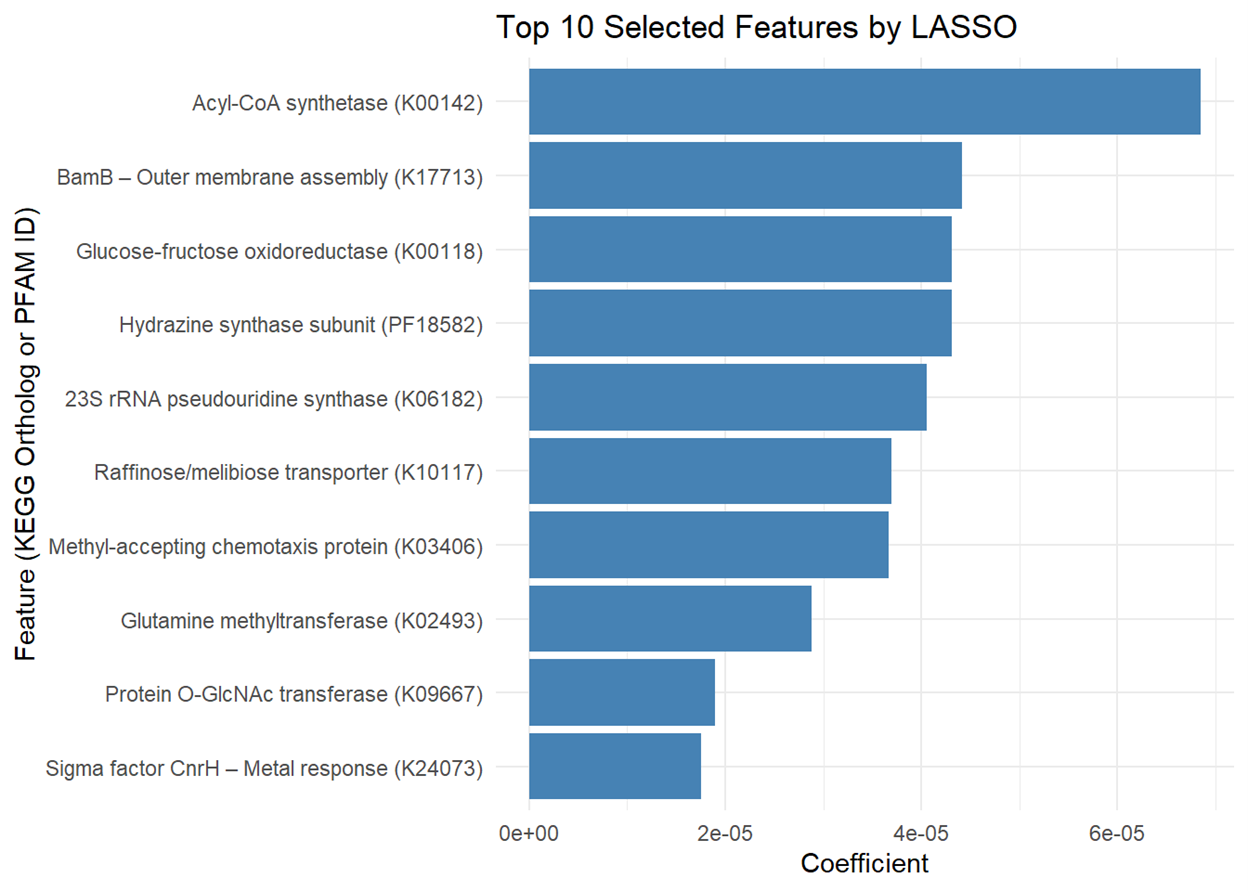
Figure 2. Top 10 Selected Features by LASSO. This figure illustrates the top microbial features identified using LASSO regression, ranked by coefficient magnitude. The features include nine KEGG orthologs and one PFAM domain, selected for their association with excess atom fraction (EAF) values. Features with larger absolute coefficients were more influential in the model, though their direct roles in microbial carbon cycling remain to be validated. Visualization generated using R.
Discussion
The results of this study demonstrate the potential use of LASSO regression in identifying microbial features associated with carbon fixation. The RMSE values for the training and testing datasets (Table 1) suggest that the model performed well, indicating that the selected KEGG orthologs and PFAM domain may play important roles in microbial carbon cycling. Specifically, the model had a training RMSE of 0.0263 and a testing RMSE of 0.0278, indicating that it effectively identified key features while maintaining reliable prediction accuracy.
Figure 2 visualizes the top 10 microbial features identified by the LASSO model, ranked by their importance in predicting microbial contributions to carbon cycling. The ranking of these features provides insight into microbial metabolism, particularly carbon fixation, the process by which microbes capture inorganic carbon and convert it into organic matter. The LASSO model identified ten microbial features, but five were selected for discussion based on their prominence in the bar plot and potential biological relevance. The approach resulted in identifying five key microbial features from the LASSO model that may influence carbon cycling. These included acyl-CoA synthetase (K00142), BamB – outer membrane assembly (K17713), glucose-fructose oxidoreductase (K00118), 23S rRNA pseudouridine2604 synthase (K06182), and a hydrazine synthase alpha subunit domain (PF18582). Acyl-CoA synthetase plays a central role in fatty acid metabolism by catalyzing the activation of fatty acids, a crucial step for their breakdown and energy production [19]. BamB is involved in the assembly of outer membrane proteins, supporting structural integrity and interaction with the environment [20]. Glucose-fructose oxidoreductase facilitates the oxidation of sugars and may contribute to microbial energy generation, particularly in fluctuating redox environments [21]. 23S rRNA pseudouridine2604 synthase introduces modifications to ribosomal RNA, which can enhance ribosomal stability and efficiency under environmental stress, potentially influencing protein synthesis in carbon-fixing microbes [22]. The hydrazine synthase domain (PF18582), found in anammox bacteria, participates in anaerobic ammonium oxidation. It catalyzes a two-step reaction: nitric oxide is first reduced to hydroxylamine, which is then condensed with ammonium to form hydrazine. This process links nitrogen and carbon cycling in oxygen-limited environments by supporting microbial energy conservation through chemosynthesis [23]. These microbial features may play important roles in metabolism, but their specific contributions to carbon cycling require further investigation. Future research incorporating KEGG Orthology (KO)-based pathway analysis could help clarify how these orthologs and domains function within broader microbial metabolic networks.
While these findings provide valuable insights, some limitations must be considered. First, this study analyzed contigs, which are short fragments of microbial DNA rather than complete genomes. This means that some connections between proteins and metabolic pathways may be incomplete. While PFAM annotations provide useful classifications of microbial proteins, they may not fully capture the diversity of microbial function. This highlights the need for more comprehensive genome sequencing and expanded annotation databases to improve future research.
To further build on these findings, future studies should explore alternative statistical learning models such as Elastic Net, Principal Component Regression (PCR), and Ridge Regression. Additionally, incorporating machine learning approaches like XGBoost, Random Forest, and neural networks could enhance predictive accuracy and refine feature selection by capturing complex relationships within the data. Furthermore, expanding the data set to include samples from various environmental conditions would help validate the generalizability of these results.
Conclusion
genomics and computational methods, with a focus on environmental exposuresThis study demonstrated that statistical methods could help predict microbial contributions to carbon cycling, even when working with incomplete genetic data. By combining protein family annotations from PFAM with LASSO regression, this research identified microbial proteins that may play an important role in carbon fixation and nutrient cycling.
Among the most significant features selected by the model were acyl-CoA synthetase (K00142), BamB – outer membrane assembly (K17713), glucose-fructose oxidoreductase (K00118), 23S rRNA pseudouridine2604 synthase (K06182), and the hydrazine synthase alpha subunit middle domain (PF18582). These microbial features are linked to a variety of metabolic processes, including fatty acid activation, membrane protein assembly, sugar oxidation, RNA modification, and anaerobic ammonium oxidation. Their selection suggests that microbial contributions to carbon cycling likely involve diverse biochemical pathways. However, the exact roles of these KEGG orthologs and the PFAM-annotated protein domain in carbon fixation remain uncertain, and further research is needed to confirm their biological significance. However, the specific contributions of these KEGG orthologs and protein domains to carbon fixation remain to be validated through experimental studies.
Although both KEGG Orthology (KO) terms and PFAM annotations were included in the dataset, most features selected by LASSO regression in this specific analysis were associated with KEGG orthologs. As a result, the analysis primarily focused on KO-based annotations to explore microbial metabolic contributions, while PFAM domain information was incorporated where available.
Beyond the scope of this study, understanding microbial roles in carbon cycling has important environmental implications. Microbial communities influence carbon sequestration, the process of capturing and storing carbon to reduce atmospheric CO₂. Refining statistical models to improve predictions of microbial contributions to carbon cycling could provide valuable insights for environmental management and climate change mitigation.
Further research should expand the dataset to include additional environmental samples, improve genome assembly techniques, and explore additional statistical and computational approaches to refine predictive accuracy. Incorporating more functional annotation tools, including KO pathway analysis, may also provide a deeper understanding of microbial metabolism beyond carbon cycling.
This study highlights the potential of integrating metagenomics, statistical modeling, and ortholog-based annotation databases to study complex biological systems, particularly microbial contributions to carbon cycling. As computational methods continue to advance, approaches like metagenomic-SIP combined with feature selection models such as LASSO will be essential for identifying novel microbial pathways involved in processes like carbon fixation, nitrogen cycling, and other ecosystem-level biogeochemical functions that influence climate and environmental health.
Acknowledgements
I would like to sincerely thank my research mentors, Dr. Elaine Luo and Dr. Wenyu Gao, for allowing me to be part of such a meaningful project. I am especially grateful to Paulo Freire, a PhD student in my lab, for his consistent and thoughtful one-on-one support throughout this work.
Many thanks to the ETHEL board for the incredible effort put into creating this journal, and to the reviewers for their insightful feedback and prompt support.
I’m also deeply appreciative of Dr. Luc Dunoyer, whose early mentorship laid the foundation for my journey in research. I truly would not be where I am today without his guidance.
Most importantly, I want to thank my parents for molding me into the woman I am today. Mom, thank you for always believing in me, even during moments when I struggled to believe in myself. Your constant support and quiet encouragement, no matter the path I chose, gave me the strength to keep moving forward. Dad, thank you for never allowing me to take no for an answer. You pushed my curiosity and problem-solving beyond limits I ever imagined, and taught me that one answer is never enough. Your influence continues to shape how I think, question, and explore the world.
With love and gratitude,
Courtney-Grace
References
- Beaulaurier, J., Luo, E., Eppley, J. M., Uyl, P. D., Dai, X., Burger, A., Turner, D. J., Pendelton, M., Juul, S., Harrington, E., & DeLong, E. F. (2020). Assembly-free single-molecule sequencing recovers complete virus genomes from natural microbial communities. Genome Research, 30(3), 437-446. https://doi.org/10.1101/gr.251686.119
- Elaine Luo, A. O., Leu, J. M., Eppley, D. M., Karl, E. F., & DeLong, E. F. (2022). Diversity and origins of bacterial and archaeal viruses on sinking particles reaching the abyssal ocean. The ISME Journal, 16(6), 1627-1635. https://doi.org/10.1038/s41396-022-01202-1
- Kieft, K., Zhou, Z., & Anantharaman, K. (2020). VIBRANT: Automated recovery, annotation, and curation of microbial viruses. Microbiome, 8(1), 67. https://doi.org/10.1186/s40168-020-00867-0
- Kirk, J. L., et al. (2015). Molecular techniques to assess microbial community structure, function, and dynamics. In Hydrocarbon and Lipid Microbiology Protocols (pp. 23–37). Springer. https://doi.org/10.1007/978-1-4419-7931-5_2
- Krause, S., et al. (2014). Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Frontiers in Microbiology, 5, 251. https://doi.org/10.3389/fmicb.2014.00251
- Kublanov, I. V., et al. (2024). Environmental activity-based protein profiling for function-driven enzyme discovery. Environmental Microbiome, 9(1), 577. https://doi.org/10.1186/s40793-024-00577-2
- LaPierre, M. J., et al. (2019). Machine learning to predict microbial community functions: An analysis of dissolved organic carbon from riverine systems. PLOS ONE, 14(4), e0215502. https://doi.org/10.1371/journal.pone.0215502
- Lemke, M. J., & DeSalle, R. (2023). The next generation of microbial ecology and its importance in environmental sustainability. Microbial Ecology, 86(4), 521-533. https://doi.org/10.1007/s00248-023-02185-y
- Liu, L., Zhou, W., & Guan, K., et al. (2024). Knowledge-guided machine learning can improve carbon cycle quantification in agroecosystems. Nature Communications, 15, 357. https://doi.org/10.1038/s41467-023-43860-5
- Lopatkin, A. J., & Collins, J. J. (2020). Predictive biology: Modelling, understanding, and harnessing microbial complexity. Nature Reviews Microbiology, 18, 507-520. https://doi.org/10.1038/s41579-020-0372-5
- McElhinney, P., et al. (2022). Interfacing machine learning and microbial omics: A promising means to address environmental challenges. Frontiers in Microbiology, 13, 851450. https://doi.org/10.3389/fmicb.2022.851450
- Nemergut, D. R., Schmidt, S. K., Fukami, T., et al. (2016). Microbes as engines of ecosystem function: When does community structure enhance predictions of ecosystem processes? Frontiers in Microbiology, 7, 214. https://doi.org/10.3389/fmicb.2016.00214
- Patil, K. R., Roux, S., & Fierer, N. (2021). Computational biology and machine learning approaches to understand host–microbiome interactions. Frontiers in Microbiology, 12, 618856. https://doi.org/10.3389/fmicb.2021.618856
- Tully, B. J., et al. (2021). Functional redundancy within the human gut microbiome. PNAS, 118(8), e2100916119. https://doi.org/10.1073/pnas.2100916119
- Zhou, Y., et al. (2022). Advances and applications of machine learning and intelligent optimization in metabolic engineering. Metabolic Engineering Communications, 15, 124-135. https://doi.org/10.1007/s43393-022-00115-6
- Neizer, Courtney-Grace. Top 10 Selected Features by LASSO. 2025. R Markdown Visualization, UNC Charlotte.
- Geneious. Assembling DNA Sequences: A Guide to DNA Sequence Assembly. Geneious, n.d. https://www.geneious.com/guides/assembling-dna-sequences
- Cui, L., Bai, L., Wang, Y., Yu, P. S., & Hancock, E. R. (2021). Fused Lasso for feature selection using structural information. Pattern Recognition, 120, 108058. https://doi.org/10.1016/j.patcog.2021.108058
- KEGG Orthology (KO) Database. Kyoto Encyclopedia of Genes and Genomes. https://www.genome.jp/kegg/ko.html
- KEGG Orthology (KO) Database. Kyoto Encyclopedia of Genes and Genomes. https://www.gejp/kegg/ko.html
- KEGG Orthology (KO) Database. Kyoto Encyclopedia of Genes and Genomes. https://www.genome.jp/kegg/ko.html
- KEGG Orthology (KO) Database. Kyoto Encyclopedia of Genes and Genomes. https://www.genome.jp/kegg/ko.html
- PFAM Database: Hydrazine synthase alpha subunit middle domain (PF18582). Pfam Protein Families Database. https://www.ebi.ac.uk/interpro/entry/pfam/PF18582/